


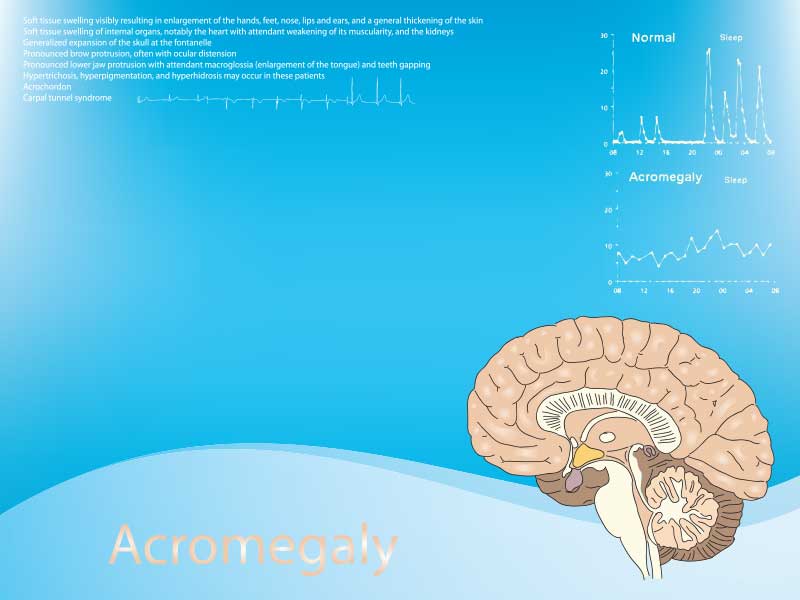
Брой 6/2021
Доц. д-р Д. Паскалев, д.м., Доц. д-р Д. Петкова
Медицински университет “Проф. д-р Параскев Стоянов” – Варна
Определение
Гигантизмът и акромегалията са ендокринни заболявания, породени от продължителна хиперсекреция на соматотропен хормон (СХ) от аденохипофизата. Абнормно високите нива на СХ водят до възникване на гигантизъм (от „gigas“, гр. – огромен, исполински) преди или по време на пубертета, а след него, при вече затворени епифизни цепки на дългите кости – до развитие на акромегалия. Гигантизмът понякога се обозначава и като „ювенилна акромегалия“ [1]. Съвременното наименование е въведено от именития френски невролог Pierre Marie (1853-1940). Терминът „акромегалия“ произлиза от гръцките думи „akron“ (крайник) и „megas“ (голям) [8,16]. През 1886 г. P. Marie описва две жени в „Салпетриерата“, Париж, като обобщава клиничната находка: „Състоянието се характеризира с хипертрофия на ръцете, ходилата и лицето, което ние предлагаме да се нарече акромегалия (acromégalie), т.е. уголемяване на крайниците…. Акромегалията се различава от микседем, болестта на Пейджет и leontiasis ossea на Вирхов“ (lion face syndrome – патологично нарастване на лицевите и черепни кости, описан от Вирхов през 1864 г. – б.а.) [цит. по 8]. По този начин името на заболяването започва с P. Marieп преди 135 години, но началото на болестта се открива далеч назад във времето.
Акромегалия и гигантизъм в Библията и древността
За пръв съобщен случай на гигантизъм следва да се приеме филистимският боец Голиат: „…от филистимския стан се изстъпи едноборец, по име Голиат, от Гет“ (Царства, I, 17:4) [2]. Пак там се съобщава, че той е “на ръст 6 лакти и педя“; ако се приеме, че един лакът е равен на 18 инча, а една педя е 6 инча, то височината на великана е около 296 см [9,11]. От библейския разказ е известно, че Голиат, облечен в тежко снаряжение, пристъпва бавно: „…филистимецът се изправи и взе да пристъпя и се приближава към Давида“ (Царства, I, 17,48) [2], а пред него крачи оръженосеца му (Царства, I, 17,38) [2]. Според някои съвременни автори по-бавното придвижване на филистимеца се дължи не само на тежкото снаряжение, но и на вероятна миопатия, известен симптом при напреднала акромегалия. Освен това те приемат, че Голиат е с нарушено периферно зрение (битемпорална хемианопсия), в резултат на хипофизарен тумор, притискащ chiasma opticum. Пак в тази връзка се посочва, че неговият оръженосец върви пред него, за да трасира посоката. По-нататък се казва, че: „Давид бързо се завтече към мястото на битката срещу филистимеца“ (Царства, I, 17,48) [2]; така лекоподвижният Давид успява да порази Голиат в челото и по-късно да го обезглави.
Допълнително някои отбелязват, че във фамилията на филистимеца има и други с гигантски ръст [9,11]. Това може да се обясни с т.нар. фамилен изолиран хипофизен аденом. Днес е изяснено, че той се дължи на мутация в AIP (aryl hydrocarbon receptor protein), който се кодира на едноименен ген, локализиран на късото рамо на 11-а хромозома. Мутацията води до либерализиране на супресорен туморен протеин, отключващ персистираща повишена секреция на СХ, по-често наблюдавана при мъже. Така прочутата библейска битка между Голиат и Давид от преди повече от 3000 години намира съвременна интерпретация [6,11]. А прекрасната мраморна скулптура на Давид във Флоренция, създадена от Микеланджело, продължава да буди възхищение. Биографът на твореца Джоржо Вазари, пише: „Няма съмнение, че след като са видели „Давид“, хората вече няма защо да гледат никоя друга скулптура, създадена през нашата или друга епоха от който и да било ваятел“ [3].
С фамилен хипофизен аденом могат да се обяснят и акромегалните черти (прогнатия и задебелени надочни дъги, arci supercilliares) при египетския фараон Птолемей I Сотер (поч. 283 г. пр. Хр.), както и у някои негови наследници, изобразени на монети от тази епоха [7]. Птолемей I е стратег на Александър Велики (356-323 г. пр. Хр.), а след смъртта му е един от диадохите (наследниците) на завоевателя, като получава Египет и основава династията на Птолемеите, чиято последна представителка е прочутата царица Клеопатра.
В римската история изпъква с огромната си фигура император Максимин I Тракиец или Трак (Gaius Julius Verus Maximinus „Thrax“ (ок. 173-328 г. сл. Хр.); той властва в периода 235-238 г. сл. Хр., а прозвището си дължи на своето родно място – Тракия. Максимин I е първият от поредицата „войнишки императори“ – наименование в историографията за обозначаване на поредица римски владетели от III в. сл. Хр., които са издигани директно от армията. Според „Historia Augusta“ императорът е висок около 238 см, употребява големи количества течности и се поти изобилно. На денариите (римски сребърни монети), както и на неговия мраморен бюст, той е изобразен с уголемена мандибула (прогнатия) и с изпъкнали надочни дъги (фиг.1 и фиг.2). Известен е и с това, че палецът на крака му е толкова голям и дебел, че носи на него като пръстен гривната на жена си. Огромният ръст, лицевите деформитети, обилното потене и полидипсията са основание да се предположи заболяването гигантизъм с акромегални черти [5,7].
През 1985 г. по време на археологически разкопки на акропол в Елефтерна, регион Ретимо на о. Крит, е разкрит гроб от VII в. сл. Хр., намиращ се под нартекса на разрушена църква. Костните останки принадлежат на индивид с неизвестен пол, починал на възраст между 25 и 40 години. Антропометричните и палеоантропологични изследвания на запазения череп показват увеличена дебелина и повишена плътност на челната и тилната кост – 90 mm, респ. 21 mm. Референтните размери за дебелина на челната кост са 79 ± 1,5 mm [4], а за окципиталната (тилна) кост – 16,1 ± 3,9 mm [18]. Турското седло (sella turcica) в открития череп е с размери: дължина 20 mm и ширина 25 mm, при референтни стойности за предно-заден размер 10,9 ± 1,8 mm [14]. Запазените фрагменти от долната челюст показват разреждане на зъбите (диастема). Костите от крайниците не са съхранени. Авторите приемат, че морфологичната находка съответства на тумор на хипофизата, предизвикал акромегалия [7].
Акромегалия-гигантизъм през XVI-XIX-ти век
Вероятно пръв състоянието в медицинската литература описва холандският лекар Johannes Wier (лат. Joannes Wierus; 1515-1588) през 1567 г. в своя труд „Medicarum observationum rararum“ („Медиицнски наблюдения върху редки заболявания“). Wier (фиг.3) изучава медицина в Париж и Орлеан и е един от първите, обявили се срещу демони и магии в живота и медицината. В „Medicarum“ той описва жена с наднормен ръст, която дефилира на сцената в различни градове срещу заплащане и така издържа себе си и своята майка. Разговаряйки с нея Wier научава, че нейните родители, както и предците ú, са били с обичаен ръст, т.е. липсва фамилност. Жената споделя, че до своята 12-годишна възраст тя е била с нисък ръст, но след 14-та си година започва бързо да израства, а менструацията ú става нередовна и окончателно спира на около 25 години. Wier съобщава, че „формите ú са неатрактивни, а тялото отпуснато“. Интересното в случая е, че авторът свързва телесните промени с настъпилата аменорея, която е известен симптом при акромегалия. Днес може да се предположи, че при жената е възникнал хипофизарен аденом с повишена СХ-секреция през пубертета [10,16].
През 1772 г. френският хирург Nicolas Saucerotte (1741-1841) съобщава пред Френската хирургична академия за клиничен случай на 39-годишен мъж с акромегалия. По-късно, през 1822 г. дерматологът Jеan-Louis-Marc Albert (1768-1837) нарича състоянието „géant scrofuleux“ („гигантска скрофулоза“), вероятно във връзка с кожните промени при това заболяване. Италианският невролог и психиатър Andrea Verga (1811-1895) нарича състоянието „prosopectasia“ („удължено лице“) и публикува клиничен случай на жена от Милано. Той съобщава, че тя има восъчна бледост и непропорционално издължено лице, за което е получила прякора „голямото лице“. След смъртта на болната от тиф (1862) Verga провежда аутопсия и установява тумор в областта на sella turcica с големина на орех, който притиска зрителните нерви; нормална хипофизна тъкан не се установява [10]. Известният италиански съдебен лекар и криминолог Cesare Lombroso (1835-1909), създател на учението за homo delictus (човекът престъпник), нарича заболяването „macrosomia“ („голямо тяло“).
Vincenzo Brigidi описва клиничната картина на известния по това време италиански артист Ghirlenzoni през 1877 г. Лекарят съобщава, че болният е със силно уголемен език, затрудняващ говора му и че лицето наподобява повече на маймуна, а не на човек; ръстът е извисен, на лице е изразена прогнатия, а бузите са удължени. След неговата смърт Brigidi открива на аутопсията „хипертрофирала хипофизна жлеза“. Той за пръв път проучва микроскопски ус 1884 г. швейцарският общопрактикуващ лекар в кантона Гларус Christian Fritsche (1851-1938) и Theodor Klebs (1834-1913), по това време патолог в Цюрих, публикуват статията „Ein Beitrag zur Pathologie des Riesenwuchses“ („Принос към патологията на гигантския ръст“). В нея те описват 36-годишен алпийски кравар с изявена акромегалия. На проведената след смъртта му аутопсия се установява увеличена тимусна жлеза и силно уголемена хипофиза в sella turcica. Авторите не дават специално наименование на заболяването [8,12].
Както вече се спомена, през 1886 г. P. Marie описва две пациентки в клиниката на прочутия Jean-Martin Charcot (1825-1893), лекувани в „Салпетриерата“. Едната е на 37 години, с уголемени длани и ходила, а лицето е с прогнатия и променено до степен, че близките не я разпознават. Болната съобщава за спряла на 25-годишна възраст менструация. Езикът е силно увеличен (макрогласия), а пациентката е с повишена жажда и често изпива чая на другите болни. Някои автори предполагат, че освен акромегалия, при нея има и стигми за безвкусен диабет (diabetes insipidus) [10]. Втората пациентка е на 54 години, със силно изпъкнали надочни дъги, широк нос и силно задебелена долна челюст. Началото на заболяването ú датира от 29-годишна възраст, когато настъпват аменорея и тежки зрителни нарушения до пълна слепота [10,12].
Към този момент P. Marie не прави връзка между телесните отклонения и евентуална хипофизна патология. През следващите години той и неговите ученици – бразилският лекар José Dantas de Souza Leite (1859-1925) и румънският Gheorghe Marinescu (1863-1938) задълбочават познанията за акромегалията. Същевременно се появяват и съобщения от други учени за болни с акромегалия и тумори на хипофизата. За дълъг период от време се водят дискусии за генезата на гигантизма и акромегалията – дали са резултат на хипофизна хипер- или хипофункция. През 1887г. немският патолог Oskar Minkowski (1858-1931) съобщава, че при всички случаи с акромегалия на аутопсия се открива уголемена хипофиза. През 1900 г. Carl Benda (1857-1932) – немски патолог и микробиолог, работещ по това време в Берлин, установява, че акромегалията е свързана с еозинофилен хипофизен тумор [16].
Продължава обаче научният спор за патогенетична връзка между двете заболявания. Marie и Souza Leite като негов ученик приемат, че това са различни болести, докато Klebs и други автори смятат, че са едно и също заболяване, но акромегалията е придобита, а гигантизмът е вроден [10,12].
Акромегалия и гигантизъм през ХХ и ХХI-ви век
През 1912 г. именитият американски неврохирург Harvey Cushing (1869-1939) издава монографията „The pituitary body and its disorders: clinical status produced by disorders of the hypophysis cerebri“ („Питуитарното тяло и неговите увреждания: клинична картина при заболяване на хипофизата“). Напълно убеден в своята теза, той още през 1909 г. получава разрешение да проучи черепа на т.нар. „ирландски гигант Charles Byrne“, известен и като O’Brien (1761-1783). Неговият скелет е съхранен от прочутият и ексцентричен хирург John Hunter (1728-1793) в музея, създаден от него. Приживе O’Brien е висок около 231 см, но анатомична дисецкия на черепа му не е провеждана. При отваряне на черепната кутия Cushing установява, че sella turcica е с размери 21х24х11 мм, което надхвърля значително референтните стойности и дава основание да се приеме хипофизна хипертрофия. През 2010 г. от зъб на гиганта е извлечена ДНК и чрез генетичен анализ е установена мутация в AIP гена. Подобна мутация е открита и при още четири негови съвременници от северноирландски фамилии с известен гигантизъм и акромегалия. Приема се, че тези хора са имали общ предшественик, живял 57-66 поколения по-рано [9,10,15]. През 1909 г. Cushing е един от първите, които постулират наличието на „hormone of growth“ („растежен хормон“), който при свръхсекреция предизвиква акромегалия. Cushing освен това привежда и собствените си наблюдения за ремисия на заболяването след провеждане на хипофизектомия. Така се поражда хипотезата за хиперфункция на аденохипофизата, водеща до клинична изява на акромегалия [12].
През 1902 г. William Bayliss (1860-1924) и Ernest Starling (1866-1927), известни английски физиолози, откриват секретина, стимулиращ панкреасната екзокринна функция; последният въвежда термина „хормон“ (от гл. движа, възбуждам). Едва през 1944 г. е получен първият говежди СХ от Choh Hao Li (1913-1987) и Herbert Evans (1882-1971) в САЩ [12]. Днес неговото изследване е обичайна клинична практика, която подпомага изследването на аденохипофизата.
Кратки биографични данни
Pierre Marie (1853-1940). Той е единствено дете на заможно семейство. Получава класическо образование с овладяване на латински и гръцки. Първоначално следва право, каквото е желанието на баща му, но по късно продължава с медицина. В периода 1883-1885 г. специализира в клиниката на прочутия Charcot, който забелязва таланта на своя ученик и го насърчава за научна кариера. През 1883 г. защитава докторат върху болестта на Базедов (Morbus Basedowi). През 1907 г. става ръководител на катедрата по патоанатомия към факултета по медицина, а от 1917г . е шеф на клиниката по неврология, на която позиция остава до 1925 г. От 1911 г. е член на Френската академия по медицина (Académie de Médecine). В медицината остава с епонимите: 1) синдром на Pierre Marie-Bamberger (хипертрофична остеоартропатия), наблюдаван най-често при белодробен карцином; Eugen von Bamberger (1858-1921) е немски клиницист, описал състоянието през 1889 г., но през следващата година P. Marie го отделя от акромегалията и 2) болест на Charcot-Marie-Tooth, т.е. прогресивна загуба на дисталната мускулатура по ръцете и краката; Howard Henry Tooth (1856-1925) е английски невролог. През 1893 г. заедно с Edouard Brissaud (1852-1909), френски лекар и патолог, създават списанието „Revue Neurologiue“ („Неврологичен преглед“), а през 1899 г. „Société de Neurologie de Paris“ („Парижко неврологично дружество“ [13].
Harvey Cushing (1869-1939) (фиг.5). Бележитият американски неврохирург е роден в Кливланд, Охайо, САЩ. Той е 10-то дете в семейството и четвърто поколение лекар. Първоначално учи в колежа на Йеил (Yale College), а по-късно завършва медицина в Харвард (1896). Насочва се към хирургията и се обучава в анестезията с етер. Той е един от първите, започнал да прилага рентгеновите лъчи за клинични нужди и през 1897 г. локализира с тяхна помощ проектилите у двама пациенти с огнестрелни наранявания. По-късно попада под благотворното влияние на прочутия интернист сър Уйлям Ослер (Sir William Osler; 1849-1919); по-късно Cushing пише биографията му „The life of Sir William Osler“, за която получава престижната литературна награда „Пулицър“ (1926). В периода 1900-1901 г. посещава редица водещи медицински центрове във Франция, Германия и Швейцария. Прави визита и на италианския лекар Scipione Riva-Rocci, като взема от него апарат за измерване на кръвното налягане и го прилага по време на операции. По това време рутинно мерене на артериалното налягане все още не съществува. През 1903 г. изнася доклад на тази тема, което го прави пионер в тая област в САЩ. След завръщането си от Европа се насочва към неврохирургията и провежда по време на кариерата си над 2000 операции за мозъчни тумори. От 1912 г. До 1933 г. работи в Бостън, където въвежда редица нововъведения – сребърен клип за хемостаза, палиативна декомпресия при неоперабилни новообразувания на мозъка и др. В периода 1908-1912 г. провежда редица наблюдения върху хипофизата и описва базофилните аденоми при болестта, носеща неговото име (Cushing disease). В по-късните си години се увлича и от история на медицината, като често посещава антикварни книжарници. Пенсионира се през 1933 г. и умира на 07.10.1939 г. от миокарден инфаркт. Наречен е „неврохирург на века“ [17].
Използвана литература
1. Орбецова М. Гигантизъм и акромегалия (Gigantismus et acromegalia), в: Ф. Николов (ред). Вътрешни болести, Лакс бук, Пловдив, 2020, 559-564
2. Библия. Синод на българската църква, София, 1982, 319-321
3. Вазари Д. Живописи на велики художници, скулптури и архитекти (Леонардо, Микеланджело, Рафаело), Изток-Запад, София, 2020, 116-117
4. Alexander S, K Rafaelis, C Gunnarsson et al. Morphological characterization of the frontal and parietal bones of the human skull, ARL-TR-7962, Mar 2017, 51-53
5. Alfaro-Martinez J. Historical figures at the office of endocrinology, Endocrinal Nutr, 2014, 61(7), 382-388
6. Berginer V. Neurological aspects of the David-Goliath battle: restriction in the giant’s visual field, IMAJ, 2000, 2, 725-727
7. Charlier P, C Tsigonaki. A case of acromegaly (Greece, 7th centery AD), Eur J Endocrinol, 2011, 165, 819-821
8. de Herder W. Acromegaly and gigantism in the medical literature. Case descriptions in the era before and the early years after the initial publication of Pierre Marie (1886), Pituitary, 2009, 12, 236-244
9. de Herder W. Familial gigantism, CLINICS, 2012, 67(51), 29-32
10. de Herder W. The history of acromegaly, Neuroendocrinol, 2016, 103, 7-17
11. Donnelly D. Hereditary gigantism – the biblical giant Goliath and his brotehrs, Ulster Med J, 2014, 83(2), 86-88
12. Mammis A, J Eloy, JK Liu. Early descripton of acromegaly and gigantism, and their historical evolution as clinical entities, Neurosurg Focus, 2010, DOI:10.3171/2010.7.FOCUS10160
13. Maranhao-Filho P, M Vincent. Who was Pierre Marie?, Arq Neuropsiquiatr, 2020, 78(7), 450-452
14. Nagaraj T, R Shruthi, L James et al. The size and morphology of sella turcica: a lateral cephalometric study, J Med Radiol Pathol Surg, 2015, 1, 3-7
15. Pears J. On the origins of pituitary apoplexy, Eur Neurol, 2015, 74, 18-21
16. Todorov S. Acromegaly, Acta Morphol Anthropol (Sofia), 2010, 16, 134-140
17. Jay V. The legacy of Harvey Cushing, Arch Pathol Lab Med, 2001, 125, 1539-1541
18. Zarghooni K, C Boese, J Siewe et al. Occipital bone thikness: implications on occipital-cervical fusion. A cadaveric study, Acta Orthop Traumat Turcica, 2016, 50, 606-609



