


Брой 11/2017
Д-р М. Сълева, проф. д-р С. Василева, д.м., проф. д-р М. Балабанова, д.м.,
доц. д-р Г. Пехливанов, д.м., проф. д-р Л. Митева, д.м.
Катедра “Дерматология и Венерология”, ККВБ “УМБАЛ Александровска”, МУ – София
Резюме
Синдромът на Киндлер е рядка генодерматоза с автозомно-рецесивно унаследяване. Въпреки че някои автори го класифицират като клиничен вариант на епидермолизис булоза, той се отличава с някои строго специфични клинични, патоморфолoгични и молекулярни характеристики. Отговорни за проявата на синдрома на Киндлер са мутации на ген, наречен KIND1 (хромозома 20p12.3), който кодира протеина Kindlin-1. Tози протеин медиира свързването на цитoфиламента актин на кератиноцитите с екстрацелуларния матрикс, чрез т. нар. “focal adhesions”.
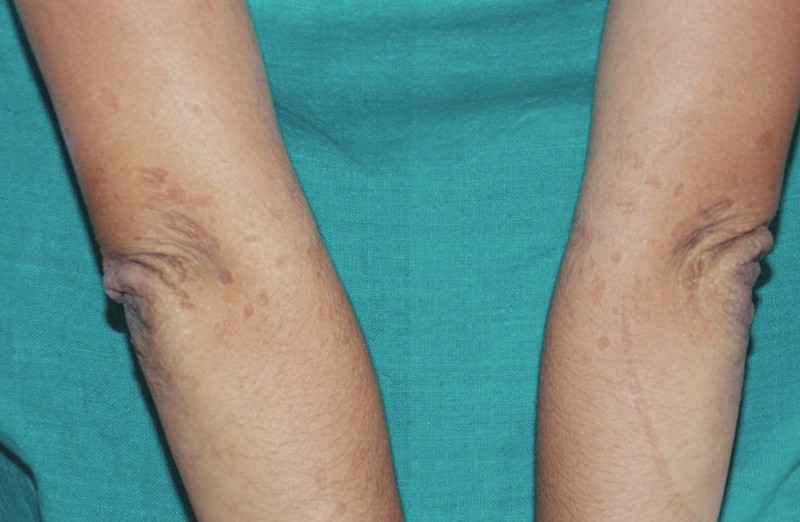
Клинично той се проявява най-често с посттравматични були, пойкилодерма, кожна атрофия, фоточувствителност, в някои случаи засягане на лигавиците.
Синдромът на Киндлер (СК) е описан за първи път през 1954 г. oт д-р Тереза Киндлер. Тя представя случай на 14-годишно момиче с поява на були по крайници, длани и ходила, фоточувствителност и пигментации по фотоекспонираните участъци (1).
СК е комбинация от черти, характерни за наследствените булозни дерматози (epidermolysis bullosa dystrophica) и конгениталните пойкилодермии (синдром на Rothmund -Thompson). Най-характерните клинични прояви на СК са поява на були по участъци, подложени на хронични травми, прогресивна пойкилодерма и кожна атрофия, основно по дорзалната повърхност на длани и ходила. Друг основен клиничен белег е изразена фоточувствителност. С напредване на възрастта, появата на були и фоточувствителност стават по-слабо изразени. Други прояви могат да са остър десквамиращ гингивит, водещ до обширни ерозии в устната кухина и загуба на зъби. Лигавиците често могат да бъдат засегнати, водейки до вагинални, уретрални и езофагеални стенози при пациенти със СК. Описано е и засягане на гастроинтестиналния тракт, като остър колит и мелена. Други прояви са псевдосиндактилия, напомняща тази при дистрофичната epidermolysis bullosa и нокътна дистрофия.
Хистологичните прояви зависят от лезията, от която е взета биопсията. Материал от булозен участък показва основата на мехура близо до дермо-епидермалната граница. На биопсия от пойкилодермична кожа се виждат хиперкератоза, епидермална атрофия и изглаждане на дермалните папили.
В дермата може да има пигментна инконтинеция, меланофаги, колоидни телца, дилатирани съдове, сходни с класическите хистологични белези на пойкилодерма.
Трансмисионната електронна микроскопия (ТЕМ) подпомага диагнозата, но често е неспецифична. Електронната микроскопия от булозно изменена кожа показва разцепване в lamina lucida, под lamina densa, или в базалните кератиноцити. Наблюдава се скъсяване на тонофиламентите на интактните кератиноцити непосредствено до булите, подобно на epidermolysis bullosa simplex.
През 2003 г. две независими групи описват гена, отговорен за СК. Генът КIND1 (FERMT1) е локализиран на хромозома 20p12.3. Този ген е човешкият аналог на гена unc-112 при Caenorhabdis elegans, кодиращ за молекула, която медиира свързването на цитосклетения филамент актин с ектрацелуларния матрикс (6).
Човешкият ген KIND1 се експресира в клетъчни култури на кератиноцити, дебело черво, бъбреци и плацента. Той кодира протеина Кindlin-1 с молекулярно тегло 77.3 кDa. KIND1 се открива в по-малки количества и в сърце, черен дроб, черва и мускулни клетки.
При новородени и малки деца поставянето на диагнозата е предизвикателство, поради сходство с клинични прояви при другите форми на epidermolysis bullosa. С нарастване на възрастта, появата на специфични белези като прогресивна атрофия, пойкилодерма, фоточувствителност и намаляване появата на були, може да насочи към поставянето на правилната диагноза. Надежден метод в диагностиката на СК е индиркетната имунофлуоресценция със специфично анти Кindlin-1 антитяло, но интензитета на оцветяването и качеството на разработваните антитела са недостатъци при този вид диагностика. Като златен стандарт за категоричното поставяне на диагнозата СК се счита мутационен анализ на KIND1.
Съществува теорията, че при пациенти със СК се наблюдава повишен риск от поява на немеланомни кожни тумори, най-често спиноцелуларен карцином, засягащ кожата на фотоекспонираните участъци и орална мукоза. В литературата са описани около 13 случаи на пациенти с доказани KIND1 мутации в съчетание със спиноцелуларен карцином.
Най-младият пациент е 16-годишно момче от Индия, проявил клиничните белези на СК – склеротични промени по крайници, pseudoainhum и пойкилодерма в областта на шията и лицето. Доказана е хомозиготна мутация в KIND1 гена, водеща до липса на Кindlin-1 в епидермиса. При него се наблюдава и бързо растящ улцериращ тумор на дясна подбедрица. Хистологично е доказан добре диференциран спиноцелуларен карцином, но с много агресивен ход, довел до летален изход на заболяването. Друг колектив описва 34-годишна жена със СК, с обширно засягане на букалната лигавица. Пациентката развива неоперабилен спиноцелуларен карцином, ангажиращ твърдото небце. Туморът е третиран успешно с лъчетерапия. Интерес представлява и българска пациентка със СК, която впоследствие развива силно инвазивен и агресивен спиноцелуларен карцином на долна устна. В литературата броят на случаите на СК в съчетание със спиноцелуларен карцином се увеличава.
Съществуват малко литературни данни за правилния терапевтичен подход при пациенти със СК.
Лечението е основно симптоматично и превантивно. Пациентите трябва да бъдат съветвани да се пазят от травми, за да се избегне появата на нови мехури. Важно е да се ограничи прякото излагане на слънце и да се използват фотопротективни средства. Болните от СК често страдат от силен пруритус, поради което е необходимо редовното хидратиране на кожата и използването на емолиенти. Поради повишения риск от развитие на спиноцелуларен карцином, е необходимо пациенти със СК да бъдат редовно проследявани. Налични верукозните лезии могат да представляват carcinoma in situ и трябва да бъдат наблюдавани и ексцизирани. Зъбният статус също трябва да се следи, като се обръща внимание на оралната хигиена.
Библиография
1. Kindler T (1954). „Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy“. Br. J. Dermatol. 66 (3): 104–11
2. Kindler syndrome: a focal adhesion genodermatosis; J.E. Lai-Cheong, A. Tanaka, G. Hawche, P. Emanuel, C. Maari,_ M. Taskesen,_ S. Akdeniz,_ L. Liu and J.A. McGrath; Genetic Skin Disease Group, St John’s Institute of Dermatology, King’s College London, Guy’s Campus, London SE1 9RT, U.K.
3. Jobard F, Bouadjar B, Caux F et al. Identification of mutations in anew gene encoding a FERM family protein with a pleckstrin homology domain in Kindler syndrome. Hum Mol Genet 2003;
12:925–35.
4. Lai-Cheong JE, Liu L, Sethuraman G et al. Five new homozygous mutations in the KIND1 gene in Kindler syndrome. J Invest Dermatol; 2007; 127:2268–70.
5. Lai-Cheong JE, Liu L, Sethuraman G et al. Five new homozygous mutations in the KIND1 gene in Kindler syndrome. J Invest Dermatol 2007; 127:2268–70.
6. Siegel DH, Ashton GH, Penagos HG et al. Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am J Hum Genet 2003; 73:174–87.
7. Arita K, Wessagowit V, Inamadar AC et al. Unusual molecular findings in Kindler syndrome. Br J Dermatol 2007; 157:1252–6.
8. Lotem M, Raben M et al. Kindler syndrome complicated by squamous cell carcinoma of the hard palate: successful treatment with high-dose radiation therapy and granulocyte–macrophage colony-stimulating factor; British Journal of Dermatology;Volume 144, Issue 6, pages 1284–1286, June 2001.
9. Has C, Yordanova I, Balabanova M, Kazandjieva J, Herz C, Kohlhase J, Bruckner-Tuderman L. A novel large FERMT1 (KIND1) gene deletion in Kindler syndrome Dermatol Sci. 2008 Dec; 52(3):209-12.
10. Cardin-Langlois E, Hanna D. Invasive squamous cell carcinoma of the hand in a patient with Kindler syndrome: Case report and literature review; MD FRCPC, Can J Plast Surg.
11. Caldeira A, Trinca WC, Flores TP, et al. A Kindler syndrome-associated squamous cell carcinoma treated with radiotherapy. Rep Pract Oncol Radiother J Gt Cancer Cent Poznan Pol Soc Radiat Oncol. 2016 Dec;21(6):532–6.
12. Natsuga K, Nishie W, Shinkuma S, et al. Expression of exon-8-skipped kindlin-1 does not compensate for defects of Kindler syndrome. J Dermatol Sci. 2011 Jan;61(1):38–44.
13. Techanukul T, Sethuraman G, Zlotogorski A, et al. Novel and recurrent FERMT1 gene mutations in Kindler syndrome. Acta Derm Venereol. 2011 May;91(3):267–70.
14. Lanschuetzer CM, Muss WH, Emberger M, et al. Characteristic immunohistochemical and ultrastructural findings indicate that Kindler’s syndrome is an apoptotic skin disorder. J Cutan Pathol. 2003 Oct;30(9):553–60
15. Youssefian L, Vahidnezhad H, Barzegar M, Li Q, Sotoudeh S, Yazdanfar A, et al. The Kindler syndrome: a spectrum of FERMT1 mutations in Iranian families. J Invest Dermatol. 2015 May;135(5):1447–50
16. Plow EF, Das M, Bialkowska K, Sossey-Alaoui K. Of Kindlins and Cancer. Discoveries. 2016 Jun;4(2).



