



Брой 1/2016
Проф. д-р И. Бойкинов, д. м. н.
AUTOINFLAMMATORY DISEASES and SYNDROMES(AIDS)
1. MULTISYSEM INFLAMMATORY DISEASES (MID)
2 WHOLE BODY INFLAMMATION (WBI)/
3. PERIODIC FEBRILTY SYNDROMЕS (PFS)
Табл.1
Списък на болестите и синдромите от групата АВЗC
ТRAPS – Tumor necrosing factor receptor associated periodic fever syndrоme; TNFR- TNF-receptor deficiency. (ММ142680)
HIDS (=Dutch синдром) – Hyperimmunoglobulin D periodic fever syndrome. (=MAPS – Mevalonate kinase associated periodic fever syndrome); (=МVKD Mevalonate kinase deficiency) (ММ260920)
MWS (=FCU)– Muckle–Wells syndrome (ММ191900)
CINCA– Chronic lammatory, neurologic, cutaneus (symptoms) and arthritis (ММ607115)
FCAS – Familial cold autoinflammatory syndrome
( =FCU – Familial cold urticaria; =CH cold hypersensitivity; =FPCE – Familial polymorphous cold eruption) (ММ120100)
РАРА – Pyogenic arthritis pustulosis acne (ММ604418))
PFAPA (=Marchall`s syndrome) – periodic fever aphtous stomatitis pharyngitis cervical adenitis syndrome
MAS – Macrophage activation syndrome
LAU (=Jabs syndrome) – грануломатозен артрит, папулозен обрив и увеит
FMF – Familial Mediterranean fever
CRMO (Mjeed syndrome) – Chronic recidivating multyfocal syndrome u PG- pyoderma gangrenosum
Behcet syndrome
Crohn disease and PPP-pustulosis palmaris and plantaris
sJIA – Systemic juvenile idiopathic arthritis
Кратки данни за АВЗ
Освен общите повтарящи се признаци – периодичен фебрилитет и свързаните с него кожни, ставни и коремни симптоми, всеки от синдромите има още някои специфики, а именно:
– HIDS – физическо и психическо изоставане, лимфаденопатия, хиперимуноглобулинемия D;
– MWS – конюнктивит, ангиоедем, конюнктивит, афтоза, ихтиоза, възможно менингит, хепатоспленомегалия;
– CINCA — прояви от първите дни след раждането, поражения на ЦНС, поражения в сензорните органи (очи, уши), умствено изоставане, епифизарни и метафизарни модификации, контрактури, възможни са хепатоспленомегалия и генерализирана лимфоденопатия;
-FCAS – симптоми в първите 1-2 часа след раждането, симптоми след излагане на студ, може и прояви от ЦНС;
– РАРА/РG – стерилен артрит с деструкции, кожни прояви на pyoderma gangrenozum;
PFAPA – афтозен стоматит и фарингит, аденопатия;
BLAU (Jabs) – грануломатозен възлест синовит, васкулит, хипертония, може и краниална невропатия, преден или заден увеит, глухота;
FMF – по-често срещан в народите около Средиземноморския басейн, освен ставни и гръдни болки, възможни са миозит, менингит, перикардит, орхит и амилоидоза;
– CRMO/PG – множествен остеомиелит, предимно на дългите кости и артрит по съседство, кожни прояви на pyoderma gangrenosum;
– Behcet – орогенитални улцерации, пустулонекротични кожни лезии;
– Morbus Crohn-PPP – грануломатозно възпаление на чревния тракт предилекционно в дисталния илиум и проксимален колон и регионален лимфаденит, коремна симптоматология;
– sJIA (системен тип на JIA) – повтарящ се фебрилитет с 2-3 пика през деня, при добро общо състояние, евентуално по-късно развитие на хроничен артрит
Терминът автовъзпаление е даден от Mc Dermot и сътрудници през 1999 г., за да се обозначи непровокирано възпаление без висок титър на автоантитела или антиген специфични Т-лимфоцити (Т-Ly). Това не са автоимунни възпаления, при които възпалението се движи (дирижира) от адаптивни имунни отговори от лимфоцитите спрямо антигенрецептори, които соматично пренастройват и мутират автовъзпалителните синдроми. Те се харатеризират с възпаление, движено от нерегулирани имунни отговори, включващи миелоидни ефекторни клетки с генетичен ред рецептори, свързани с патогенни агенти и опасни сигнали. (19).
През 1999 г. са идентифицирани генетичните причини на две моногенни автовъзпалителни болести MEFV за FMF (Фамилна Средиземноморска треска) и TNFSF1A за болестта TRAPS (Tumor Necrosing Fsctor Receptor Associated periodic Fever). Чрез изследването GWAS (genom wide assocated studies) се прави непрекъснато опит за идентифициране генетичните причини на останалите автоимунни възпалителни болести и синдроми. (19).
Като автовъзпалителни заболявания са дефинирани повтарящи се възпалителни явления без доказан инфекциозен агент. Не са доказани нито автоантитела, нито антиген специфични Т-лимфоцити. Около 14 заболявания и синдроми, по-голямата част от които вродени, се очертават като продукт на вродената инстинктивна имунна система (innate immune system – InnIS), в която са въвлечени различен брой генни мутации и един интрацелуларен голям протеинов комплекс (ГПК).
J.Roth доказа 3 свързани с калций провъзпалителни молекули от ГПК , които играят важна роля в IniIS . Те се експресират и секретират от фагоцитите и се доказват в кръвта и синовиалната течност на възпалените стави.
Установени са отговорните гени на почти всички заболявания от тази група и всеки от тях кодира определена област (домен) от ГПК. Всеки домен притежава свойството да влиза във взаимодействия с всички останали домени с изход в CARD, NF-kP или App домените (предстаени на фиг 1). Последните са въвлечени по един още ненапълно уточнен механизъм в развитието на възпаление или апоптоза.
Възпалението се представя от увеличени CRP, СУЕ и проинфламаторните цитокини IL-1. TNF-α , IL-18.
Отделните болести и синдроми са посочени в статията.
Направен е обстоен обзор на настоящите знания за тази интересна група болести и синдроми.
При лечението на АВЗС антибиотиците и НСПВС не оказват ефект, такъв оказват само ГКС и биологичните средства като блокаторите на TNF-alpha и IL-1.
Това е група от заболявания и синдроми, характеризиращи се с интермитентни самоограничаващи се възпалителни периоди. Повечето имат начало в ранната детска възраст, дори веднага след раждането и се очертават като oсобени синдроми с клинична характеристика, включваща общи споделени симптоми от страна на стави, кожа, коремни органи + периодичен фебрилитет. Всеки от тях има и специфични прояви, които му придават индивидуалния фенотип. Това е наложило използването на названия, съставени от инициалите на клиничните симптоми .
Етиологията и патогенезата са обект на голям интерес и интензивни изследвания. В патогенезата се очертава ролята на една вродена инстинктивна имунна система – ВИИС. Тя е разпозната по способността й непосредствено и бързо да реагира на бактериемия и на липополизахариди (2-4 ).
От редица изследвания е установено, че за ВИИС оперира чрез един голям ендоплазматичен протеинов комплекс – (ГПК) (1). Той е съставен от по-големи и по-малки комплекси и протеинни групи (с общо название “домени”), кодирани и активирани от съответни мутантни гени. Протеините на ГПК имат свойството да взаимодействат помежду си и това взаимодействие играе важна роля в живота на клетката и медиира заболяванията от тази група.(1)
Гените могат да имат различен брой вродени мутации, като активирането на определена мутация може да инициира определен ход и начин на взаимодействия между кодираните от нея протеини с останалите протеини на ГПК. Това е субстратът на активираната ВИИС, с краен резултат развитие на особен тип възпаление или апоптоза по един още неуточнен напълно механизъм (фиг. 1).
В патогенезата на тези заболявания и синдроми се очертава ролята на една вродена инстинктивна имунна система (наречена още древна) – ВИИС (InnIS). Тя е разпозната по способността й непосредствено и бързо да реагира на бактериемия и на липополизахариди (2-4 ).
От редица изследвания е установено, че за ВИИС от основно значение е един голям ендоплазматичен протеинов комплекс – (ГПК) (фиг.1). Цитоморфологично той се представя от една своя част като цитоплазматичен агрегат, съставен от протеините на ASC(=NSC) u pyrin. При флуоресцентно маркиране и боядисване с rhodamin той се вижда като двойно контурирано петно (specks), оцветено в жълто (oт АSC) и червено (от pyrin) (фиг.2). Specks се формира основно от АSC и се смята за предвестник на апоптозата, докато pyrin изглежда да противодейства на АSC (8, 9).
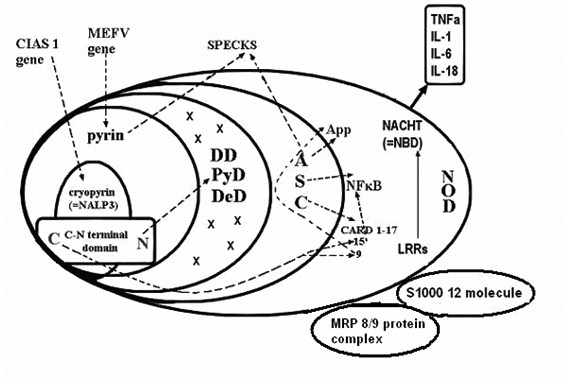
Фиг. 1 Голям ендоплазматичен протеинов комплекс (ГПК).
Състав:
Cryopyrin (=marenictrin, =NALP3) – linked cold temperature exposure to inflammation (protein product na CIAS1 gen)
Pyrin – protein (90 aminoacids) product na MEFV gen
N-C terminal domen
DD (=PyD) – deat domen related structure
DeD – death effector domen
ASC (=NSC) – apoptosis associated speck-like protein
App – Appoptosis domen
NF-B – Nuclear factor kappa B , Transcription factor involved in initiation and resulation of the inflammatory response
CARD 1-17 – Capsase recruitment and activating domen
NOD (=NACHT) – Nucleotid binding oligomerization domen
NOD1 (=CARD4); NOD2 (=CARD15)
NACHT (=NBD) – Nucleotid binding domen
CIAS1-gen – Could induced autoinflammation
MEFV–gen of FMF
Specks – protein aggregate from pyrin and ASC(NSC)
LRRs – Leucin rich repeats domen
N.B. Названията съответстват отчасти на функцията или на състава им, или са случайни.
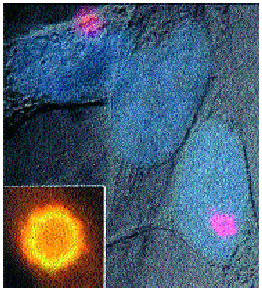
Фиг..2. Specks – цитоплазмена структура, съставена от ASC и pyrin протеините, разпознат чрез двойно маркирана имунофлуоресценция. Показани са две клетки с specks с червена оцветка (тук той е визуализиран чрез боядисване на pyrin-протеина с rhodamine). Малката фигура, включена в картината, показва силно увеличен specks. Жълтият ореол около кръглата структура е получен от едновременното наличие на ASC (зелено) + pyrin (червено).
На фиг.1 е представена сложната структура и съотношенията между домените и протеинните групи на ГПК. Cryopyrin (=marenoctrin) доменът се включва в pyrin domen, а той – в PyD (=DD) и в NSC (=ASC). Различните изследователи означават отделните домени и групи с различни названия, които се явяват синоними и ние ги поставяме в скоби: PyD (=PAAD,=DAPIN); NOD1 (=CARD4), NOD2 (=CARD15), NOD-IPaf (=CARD12) и т.н.
PyD доменът съдържа структура, имаща отношение към програмираната клетъчна смърт (DD – death related structure) и редица по-малки структури с неясна функция (х). Cryopyrin и pyrin имат линеален субдомен с С и N терминал. N терминалът взаимодейства и активира DD, а С терминалът – CARD15 (виж фиг.1). Следват домените NACHT (NBD) и този на протеини, богати на левцин- LRRs .
ASC (=NSC) чрез своите 3 ефекторни субдомена NF-кB, App и CARD участва в два важни изходни процеса – възникване и развитие на възпаление (NF-кB) или апоптоза (App). CARD може да активира или възпалението, или апоптозата, взависимост от това дали активирането преминава през CARD15 или CARD9 (фиг.1). Кога и при какви обстоятелства се активира всяка от трите ефекторни протеинни групи не е напълно изяснено. Значението на активирането на капсазите на CARD също така подлежи на уточняване.
Непосредствени медиатори на възпалението и тъканното увреждане изглежда да са протеините и протеинните субкомплекси HNGB1, HsPs и S100Ps, обединявани като DAM-протеини (DAMPs). Намиращи се най-вероятно в периферията на ГПК в домените NACHT и NOD (фиг.1), те медиират активирането на имуноцитите, провъзпалителните цитокини като TNF, протромботичните молекули и matrix metalloproteinase 13 (в моноцитите) (16 ). Силно експресирани на левкоцитите и освободени в извънклетъчното пространство, те предизвикват експресирането на адхезионни молекули на епителните клетки, при което увеличават пермиабелитета през ендотелите.
DAMPs имат двойствено действие: вътре в клетката играят роля в клетъчната хомеостаза като свързват калциевите йони и стабилизират хроматина. Силно експресирани на левкоцитите и освободени в междуклетъчното пространство и в кръвното русло (18), те реализират своята медиаторна роля на възпалението.
S10012 И MRP8/14 (S100A8/9) СА ПРОВЪЗПАЛИТЕЛНИ ПРОТЕИНИ.
С100А8 и С100А9 (=MRP8/14) много силно експресирани на моноцитите и неутрофилите, пристигнали в огнището на възпалението, бързо се освобождават. С100А8 и С100А9 проявяват проинфламаторния си ефект и чрез епителните клетки, като ги стимулират да експресират адхезионни молекули и да увеличат пермеабилитета.(16).
На фигура No1 са посочени и основните гени, които кодират протеинните домени и субдомени. Съществуват различни възможности. Aктивирането може да започне от един или друг мутантен ген и да последват активиращи взаимодействия между домените на ГПК, които следват различни посоки и пътища до ефекторните субдомени (показани на фигурата със стрелки.)
ГПК трябва да се представя не в плоскост, както е на фиг.1, а пространствено, като формация с отделни взаимнопроникващи се пластове.
Както личи от фиг. 1, ГПК е съставен от по-големи и по-малки комплекси и протеинни групи (домени), кодирани от съответни гени, основни от които са CIAS1, MEFV, CARD, NOD и др. (фиг.1). Протеините на ГПК имат свойството (когато са активирани от съответни мутантни гени) да взаимодействат помежду си и това взаимодействие играе важна роля в живота на клетката и медиира симптомите на заболяванията от тази група.
Гените могат да имат различен брой вродени мутации, като активирането на определена мутация може да инициира определен ход и начин на взаимодействия между кодираните от нея протеини с останалите протеини на ГПК. Това е субстратът на активираната ВИИС с краен резултат развитие на особен тип възпаление или апоптоза по един още неуточнен напълно механизъм. Активирането преминава по различни пътища на вътрешни взаимодействия на домените и финишира в ефекторните домени ASC (=NSC), CARD, NF-kappaB, и App, които са въвлечени в развитието на възпаление и/или на апоптоза – два процеса в променлив баланс във времето.
Проинфламаторните протеини MRP8/14 (=S100A8/9) и S100A12 са свързани с големия протеинов комплекс (ГПК) (фиг.1) и играят важна роля в развитието на възпалителния процес (литература PRES Ljubljana, 2013 Poster No 2216).
Както при другите познати типове възпаление (клетъчно, имунокомплексно и IgE- медиирано) и тук е налице свръхпродукция на проинфламаторни цитокини IL-1, IL-18, TNFα, IL-6 и острофазови протеини.
Агентите, които провокират активирането на InnIS остават недоловими. Предполага се ролята на незначителна, пренебрегвана бактериемия, коменсална гърлена флора или малки стресови моменти. Сензори на провокиращите агенти се оказват преди всичко основните гени, наскоро доказано специално за NOD1 и NOD2 (7).
Клинично този генетично обоснован тип възпаление се проявява в периодичен фебрилитет, асоцииран с още 3 прояви – кожни, ставни и коремни, повтарящи се при всички синдроми от групата на AВЗ. Останалите специфични за даден синдром симптоми, които могат евентуално да се повторят при някои от останалите синдроми, (виж табл. 2) се определят най-вероятно от началния активиран ген (определена негова мутация) и от посоките и пътищата, по които минава активирането на взаимодействията между отделните домени. Това обяснява възможността активирането на един и същи мутантен ген да доведе до реализирането на два или повече синдрома – например BLAU u Crohn при NOD2 (=CARD15) мутантен ген и FCU u CINCA при CIAS1 мутантен ген. Друга причина може да бъде активирането на два или повече мутантни гени. С активирането на cryopyrin домена са асоциирани MW, FCAS и CINCA синдромите.
Следователно, активирането на ГПК може да се реализира под влияние на мутациите на един или повече от гените, кодиращи протеините на ГПК, но също така и на мутациите на гените на провъзпалителните цитокини и техните рецептори, както например е случаят при TNFR–синдрома.
Една от най-странните прояви на болестите от тази група е повтарящият се фебрилитет, по време на който се появяват или засилват всички симптоми на заболяването.
Нормално в организма действа вътрешен часовник на денонощния ритъм на концентрацията на мелатонина (произвеждан от питуитарната жлеза и увеличаващ се през нощта) и на вероятно свързаните с него ден-нощ колебания на телесната температура. Часовникът е локализиран в предните хипоталамични ядра. Циркадианното му функциониране се основава на действието на една невроендокринна-имунна ос: хормон – цитокин–хипоталамус. Пусков момент е смяната на светлина с тъмнина. Koгато се касае за периодика във времето, (не денонощна) най-вероятно се касае за ролята на друг/и пусков/и момент/и (стрес, пренебрегвана бактериемия или лека инфекция). Затова няма фиксирана периодика на фебрилните пристъпи.
При периодичните фебрилни синдроми (ПФС) очевидно настъпва разстройство на функциониране на вътрешния часовник и преминаване на нова програма на действие и евентуално при друг пусков фактор. Особен интерес представляват изследванията по отношение на проинфламаторния цитокин, който е от значение при даден периодичен фебрилен синдром (11). При MW синдрома е установено точно съвпадение на фебрилните атаки и уртикарията с увеличена концентрация на IL-6.
На табл. 2 са представени относително специфичните симптоми нa Behcet синдромa и при кои други синдроми те също се срещат.
Клинични манифестации Синдроми
Улцеро-афтозен стоматит HIDS
Генитални улцерации HIDS
Акнеформени лезии PAPA
Патергична реакция PAPA
Увеит и други очни BLAU, CINCA,TRAPS
Менингоенцефалит FMF, CINCA
Oрхит-епидидемит FMF
Периодичен фебрилитет, артрит, кожни, коремни – при всички синдроми
Интересен е също фактът, че един и същ синдром се развива при отделните популации от активирането на различни мутации на гена, отговорен за заболяването.
NOD е домен, чиито молекулярни детайли не са установени. Той включва редица други домени: CARD, NF-кB, App, NACHT(=NBD) и LRRs.
NOD1 u NOD2 cа домени, на които много изследователи поставят равенство c CARD: NOD1=CARD4, NOD2=CARD15 u NOD-Ipaf=CARD12. За CARD се знае, че съдържа 17 субрегиона – капсазе 1-17, които имат различна функция при активирането им.
Взависимост от вида на мутацията на NOD-гена, активирането може да мине през взаимодействия на 5-те субдомена на NOD по различен начин. През CARD15, NACHT (=NBD) към NF-кB се реализира възпаление, а през CARD9 и изключване на LRRs се реализира апоптоза. LRRs инхибира NACHT(NBD) ! (виж фиг.1).
NOD, CARD, NACHT (=NBD), NF-кB, App са домени, съдържащи групи протеини, тясно свързани и взаимнопроникващи едни в друг, кодирани от близко стоящи гени. Hай-нови изследвания (М.Albrecht et al – 2003) говорят за тясна локализация, дори за идентичност на тези гени. Това обяснява защо някои автори поставят равенство между протеинните групи (домени), кодирани от тях. Това загатва за една интегралност в патогенезата на всички синдроми. Всъщност, най-вероятно се касае за варианти на единния ген (ако се приеме съществуванието на такъв), които кодират протеини със специфични функции и специфични взаимодействия, оттам и специфичните характеристики на отделните синдроми. Друго алтернативно обяснение е, че спецификите на отделните синдроми се обуславят от ролята на една отделна определена мутация на гена (един ген може да има много мутации) и/или от различните пътища на вътрешните взаимодействия между протеините на ГПК (както бе указано по-горе) или на едновременно активиране на няколко варианти на гена.
Заключение
Касае се за заболявания с недоказана етиология и неизяснена напълно патогенеза. Клиничната симптоматика отговаря на високо възпаление при силно повишени провъзпалителни цитокини в кръвта и урината. Общото в клиничната картина на синдромите от тази група е периодичният фебрилитет, ставните и кожните прояви. В хода на болестта съществува склонност за развитие на амилодоза. Фебрилните периоди са рецидивиращи и самолимитиращи се. Всички симптоми, включително и специфичните, изчезват бързо или бавно, или силно атенюират след температурните атаки.
В Германия е изработена следната дефиниция на “Периодичните фебрилни синдроми”(ПФС): Начало в детската възраст <=16 год. с > от 3 фебрилни пристъпи с типична клинична картина, високи стойности на показателите на възпаление и асоциация с определени генни мутации.(5).
В патогенезата на тези синдроми е доказана ролята на мутации на различни гени като CIAS1 (при FCU, MWS и CINCA синдроми), CARD (при MVKD синдром), NOD (при BLAU синдром), MVK (при HIDS синдром) и др. Те кодират протеините от един голям протеинов комплекс, който стои в основата на ВИИС. Подробностите в механизмите, по които активирането на ВИИС води до увеличено производство на IL-1, TNFα и другите инфламаторни цитокини, са в процес на изясняване. Мутации и на други гени могат да имат същия ефект.
Така например, мутантен TNF ген (намиращ се в 6-та хромозома в близост до HLA), който води до дефектна регулация в продукцията на TNFα, може да предизвика включването на ВИИС, възпалителен отговор и периодичен фебрилитет (например синдрома TRAPS) (10).
Организирано e събирането на случаите от даден синдром в един европейски център за системно обработване, изследване и уточняване на диагностичните критерии.
Обсъждане
Синдромите от тази група носят белезите на възпалението – фебрилитет, увеличениe на полимофоядрените левкоцити, острфазовите белтъци (СRP) и инфламаторните цитокини (IL-1, Il-15, IL-18, TNFα) в кръвта. По механизъм на възникване те не отговарят на познатите клетъчно, имунокомплексно или IgE-медиирани възпаления1. Няма в основата имунен конфликт между екзо или ендогенни антигени и имунната система. Няма разпознат антиген, нито антитела във висок титър или въвличане на антиген-специфични Т-лимфоцити.
В патогенезата им играят роля вродени мутации на гени, кодиращи продукти, които стимулират синтезата на инфламаторните цитокини. Механизмът на тази стимулация не е напълно изяснен, но изследванията са показали голям напредък.
Патологичният биологичен процес не отговаря на класическата представа за възпаление (чисто имунологично дефинирано). Има редица общи особености в клиничните изяви: начaлото на повечето от заболяванията е в кърмаческата и ранна детска възраст, фебрилитетът е рецидивиращ, проявите от страна на кожата и ставите са налице при всички синдроми и кореспондират точно с фебрилните пристъпи. Както едните, така и другите, са самоограничаващи се (този момент остава най-странен и необясним), не се излекуват от НСПВС, циклофосфамид и mycophenolat mofetil, непълно, частично пристъпите се влияят от ГКС (големи дози), a се поддават на лечение, макар и непълно, с биологичните средства kinеret, infliksimab, etanercept и humira.(12,13,14).
При заболяванията от тази група с по-късно начало може да се предполага придобита мутация на респективните гени!
Литература
1. Kastner DL. Hereditary periodic fever syndromes. Hematology (Am Soc Nematol Educ Program) 2005:74-81
2. Stojanow S. Kastner DL. Familial autoinflammatory diseases: genetics, pathogenesis treatment. Curr Opin Rheumatol. 2005;17:586-599
3. McKann L, Hasson N, Woo P. Favourable preliminary results for the use of anti TNF therapy in the treatement of the periodic fever syndromes, TRAPS and HIDS: experience in 7 children. XII European Pediatric Rheumatology Congress, Versailles, 15-18.09.2005. Clin Experim Rheumatology, 23,3:P197, S54.
4. Livermote P, WooP. Dramatic response to anakinra in 3 groups of youg onset multisystem inflammatory diseases, but infection is a risk. XII European Pediatric Rheumatology Congress, Versailles, 15-18,09, 2005. Clin Experim Rheumatology 23,3:p-194,S53
5. Lanka E, Neudorf U, Huss K et al. Hereditare periodische Fibersyndrome (HPF) – eine Inzitenzmittelung in Deutschland. Z Rheumatologie, 2005, 64, 1:FKJ-3-4
6. Kummerle-Dreschner JB, Tzaribachev N, Dannecker GE et al. Anakinra zur Behandlung des Mucke-Wells syndroms – eine serie von vier Fällen. Z Rheumatologie, 2005,64,1: FA1-3,1/5
7. Muratori F, Cortis E,Urbano L e al. Abnormal regulation of inflammatory cytokine production in PAPA syndrome. XI European Pediatric Rheumatology Congress, Kosice, Slovakia, 9-12.09.2004. No 164, S538
8. Richards N, Shander P, Diaz A et al. Interactions between pyrin and apoptotic specks protein (ASC) modilates ASC – induced apoptosis. J Biol Chem . 2001, 276:39320-9
9. Masumoto J, Taniguchi S, Ayukawa K et al. ASC a novel 22kDa protein aggregates during apoptosis of human promyelocytic leucenia HL 80 cells. J Biol Chem 1999, 27433835-8
10. Liz-Grana, Gomez-Reino Carnota JJ. Tumor necrosis factor. Genetics, cell action mechanism and involvement in inflammation. Alergol Immunol Clin. 2001,16:140-149
11. Gerbig AW, Dahinden CA, Mullis P et al. Circadian elevation of IL-6 levels in M-W syndrome: disorder of the neuro-immune axis. Q J Med, 1998, 91:489-492
12.McKann L, Hasson N, Woo P. Favourable preliminary results for the use of anti TNF therapy in the treatement of the periodic fever syndromes, TRAPS and HIDS: experience in 7 children. XII European Pediatric Rheumatology Congress, Versailles, 15-18.09.2005. Clin Experim Rheumatology, 23,3:P197, S54.
13.Livermote P, WooP. Dramatic response to anakinra in 3 grous of youg onset multisystem inflammatory diseases, but infection is a risk. XII European Pediatric Rheumatology Congress, Versailles, 15-18,09, 2005. Clin Experim Rheumatology 23,3:p-194,S53
14. Kummerle-Dreschner JB, Tzaribachev N, Dannecker GE et al. Anakinra zur Behandlung des Mucke-Wells syndroms – eine serie von vier Fällen. Z Rheumatologie, 2005,64,1: FA1-3,1/5
15. Muratori F, Cortis E,Urbano L e al. Abnormal regulation of inflammatory cytokine production in PAPA syndrome. XI European Pediatric Rheumatology Congress, Kosice, Slovakia, 9-12.09.2004. No 164, S538
16 .Roth J. Role of innate immunity in control of autoimmunity. Annals of Rheumatic diseases.2006, Vol.65,suppl.11, SP0068, P24
17. Potempa J. The coruptionof innate immunityby bacterial proteases. Annual Eular Cngress London, 25-28 May 2011. Annals of Rheumatic diseses. Abstracts SP0027, P.9
18. Holzinger D. S100 Biomarkers as tools for guiding clinical practice. Annual Eular Cngress London, 25-28 May 2011. Annals of Rheumatic diseses. Abstracts SPOO22, P8
19. Romers. The expending spectrum of autoinflammation. Annual European Congress of Rheumatology, Madrid, Spain, 12-15 VI, 2013, Abstract SPOOO9



