



Брой 1/2017
Д-р Д. Стоянова, доц. д-р Н. Стоева
Отделение по пневмология, МБАЛ „Токуда Болница София“ – София
Резюме: Идиопатичната белодробна фиброза (ИБФ) е хронична прогресираща болест с неизвестна етиология, с необратимо нарушение на белодробната архитектоника, спад на форсирания витален капацитет (ФВК) и нарушения в кръвно-газовата обмяна.
Ключова роля за започването и поддържането на фибротичния процес има продължителната микроувреда на епитела на дихателните пътища и непълното му възстановяване, включващо участието на редица възпалителни и фибротични фактори.
Проведените досега клинични проучвания показват, че лечението с медикаментите нинтеданиб и пирфенидон забавя леко спада на ФВК, намалява честотата на екзацербациите и повлиява положително смъртността.
Идиопатичната белодробна фиброза (ИБФ) е прогресиращо, необратимо, хронично, рядко и накрая фатално заболяване1,2.
Тя е част от групата на интерстициалните белодробни болести, представляващи хетерогенни паренхимни белодробни болести, характеризиращи се с дифузно, хронично възпаление и фиброза (с различна степен на изразеност) на белодробната съединителна тъкан, главно в най-периферния и нежен интерстициум – този в алвеоларните стени. Често са засегнати и периферните въздушни пътища и съдовете около тях3. Групата на интерстициалните пневмонии (ИП) е една от основните, характеризиращи се с рестриктивен тип нарушение, абнормна газова обмяна и характерен рентгенов образ. Обединени са в група поради сходни клинични белези, симптоми, рентгенологични и патофизиологични промени. Интерстициалните белодробни болести (ИББ) заемат около 15% от неинфекциозните белодробни болести. Според последната класификация от 2013 г. на Американската торакална асоциация и на Европейската торакална асоциация ИП се групират на база причина за възникване (идиопатична срещу вторична), доминиращ морфологичен модел, тежест на протичане (обратими, стабилни, прогресиращи), прогноза и терапевтично поведение4. Така прегрупирането може да се представи по няколко начина в зависимост от критерия за обединяване – на базата на епидемиологичната характеристика (табл.1), на базата на хистологичната характеристика (табл.2) и на базата на далечната прогноза (табл.3).

Идиопатичната белодробна фиброза (ИБФ) представлява рядка (4,6-16,3 на 100 000 човеко-години) специфична форма на хронична прогресивна фиброзираща пневмония с неизвестен произход, развиваща се при възрастни лица5-8. Рентгенологично и хистологично тя съответства на обикновената интерстициална пневмония9. Независимо от съвременната анти-инфламаторна терапия, болестта има прогресивен, непредсказуем ход, като промените са необратими и водят до летален изход, като продължителността на живота от времето на поставяне на диагноза е средно между 2-5 години 10.
Патогенеза
В патогенезата на белодробната фиброза участие вземат много фактори – генетични, увреждащи фактори от околната среда, коагулационната каскада, процесите на стареене. Типичната характеристика на ИБФ включва пръснати фибробластни огнища (агрегати от фибробласти и миофибробласти), граничещи с разрушен белодробен паренхим. Фиброзата в белия дроб е резултат от възникването на множество зони на епителна микроувреда. След стартиране на процеса увреденият епител навлиза в състояние на свързан със стареенето секреторен фенотип, при който се произвеждат възпалителни и фибротични фактори, подпомагащи оздравителните процеси в белия дроб11.
Клинична характеристика
Ранните симптоми, характерни за ИБФ, са:
- прогресивно засилващ се задух при физическо усилие, а впоследсвие и в покой;
- упорита суха кашлица;
- засягане предимно на мъже;
- засягане предимно на пациенти > 45 години;
- отпадналост;
- болки в мускули и стави;
- отслабване на тегло;
Симптомите много често се интерпретират като свързани с възрастта, хронична застойна сърдечна недостатъчност, хронични белодробни заболявания (бронхиална обструкция или паренхимни нарушения), белодробна грануломатоза, ГЕРБ, като поставянето на точна диагноза понякога отнема месеци, дори години.
Физикален статус:
- Бял дроб: Двустранни базални крепитации в края на инспириум („велкро“ крепитации – звук, наподобяващ разлепване на велкро лента)
- Сърце: Акцентуиран P2 (пулмонална хипертония)
- Крайници: Барабанни пръсти
Функционално изследване на дишането
Функционалните изменения при ИБФ са като при рестриктивно заболяване, с намаляване на дифузионния капацитет, белодробния обем, къмплайнса, като 6MWT (6 minute walking test, 6-минутен тест при ходене) е лесен и точен тест за проследяване прогресията на заболяването.
За прецизиране на диагностичния процес са приети образните методи на изследване и инвазивна диагностика, а именно високорезолютивна компютър-томография (HRCT) и видеоасистирана торакоскопия (VATS) или други методи за осъществяване на отворена хирургична биопсия с хистологична верификация 12,13.
Образни изследвания
Нормалната рентгенография не може да изключи ИБФ. Термини от разчитането на рентгенографията, които се свързват с ИБФ са интерстициални промени, фиброза, засягане предимно на долни дялове.
Компютърната томография (КТ) има основна роля при поставяне на диагнозата ИБФ.
Обикновено КТ находка е характерна и достатъчна за поставяне на диагнозата, без да се налага провеждане на отворена белодробна биопсия, при изключване на известни причини за възникване на фиброзни изменения в паренхима на белите дробове като хронична прахова експозиция или такава на различни биологични и химични агенти, съпътстващи заболявания на съединителната тъкан, лекарствени взаимодействия и други14. КТ образът е както при обикновена интерстициална пневмония (UIP) и се представя с характерни рентгенови белези. Образните критерии включват наличието на двустранни, предимно базални, субплеврални ретикуларни засенчвания. Наличието на промени по типа на „пчелна пита”(honeycombing) е това, което определя дефинитивно диагнозата. Промените тип „пчелна пита” се представят като групирани псевдокистични зони с диаметър от 3-5 мм с неравномерно дебели стени. Промените тип „пчелна пита” имат позитивна предиктивна стойност при поставяне на диагнозата 90-100% (фиг. 4) 15. Белодробната фиброза се представя на КТ с ретикуларни промени, локализирани предимно периферно и сублеврално. Апикалното или централно разпределение на промените обикновено предполага вторично ангажиране след хиперсензитивен пневмонит (ХСП) или системно заболяване на съединителната тъкан. В по-авансиралите случаи се среща образът на тракционни бронхиектазии и архитектурна преустройка на паренхима16.
На базата на комбинацията от белези, ревизираните диагностични критерии на ESS/ERS/JRS/ALAT от 2013 г. определят данните от високо-разделителна компютърна томография (ВРКТ) като „типични за ОИП”, „възможна ОИП” и „непокриващи критериите за ОИП”
Хистология
Хистологичният модел на фиброза е като при обикновена интерстициална пневмония (ОИП; usual interstitial pneumonia, UIP), което се изисква за диагнозата ИБФ. Определение: ОИП е хистологичен модел на ИББ, при което се наблюдават двустранни, дифузни, паренхимни белодробни увреждания.
За подпомагане на диагностицирането на интерстициалните белодробни болести през 2011 г. са приети хистопатологични критерии за ИБФ (да са налични и четирите) и те са следните:
1. Доказване на изразена фиброза/промяна в строежа, със или без изменения тип „пчелна пита”, с предимно субплеврално/парасептално разпределение;
2. Наличие на неравномерно засягане на паренхима от фиброза;
3. Наличие на фибробластни огнища;
4. Липса на изменения, характерни за други болести18 (хиалинни мембрани; организираща пневмония; грануломи; изразен интерстициален възпалителен инфилтрат; предимно бронхиолоцентрични изменения; други характеристики, предполагащи алтернативна диагноза -трябва да е наличен поне 1 от 6-те критерия за изключване на диагнозата).
Съществуват също така хистопатологични критерии за вероятна и възможна ИБФ.
Инвазивни диагностични процедури
Диагнозата на фиброзиращите интерстициални белодробни болести (ФИББ) е стъпаловиден процес. Инвазивните процедури влизат в съображение само когато клиничните данни, лабораторните тестове и HRCT са с недостатъчна диагностична стойност19,20. Пулмолозите разполагат с широк набор от инвазивни процедури, включващ бронхоалвеоларен лаваж (БАЛ), ендобронхиална биопсия,трансбронхиална белодробна биопсия (ТБББ) с конвенционална щипка или крио-трансбронхиална белодробна биопсия (крио-ТБББ), сляпа или трансбронхиална аспирационна биопсия (ТБАБ) с ЕБУЗ (ендобронхиален ултразвук), ЕУЗ (ендоскопичен ултразвук), трансторакална биопсия (под КТ или ехографски контрол) и видеоасистирана торакоскопия (VATS)21-24.
Диференциална диагноза на идиопатичната белодробна фиброза
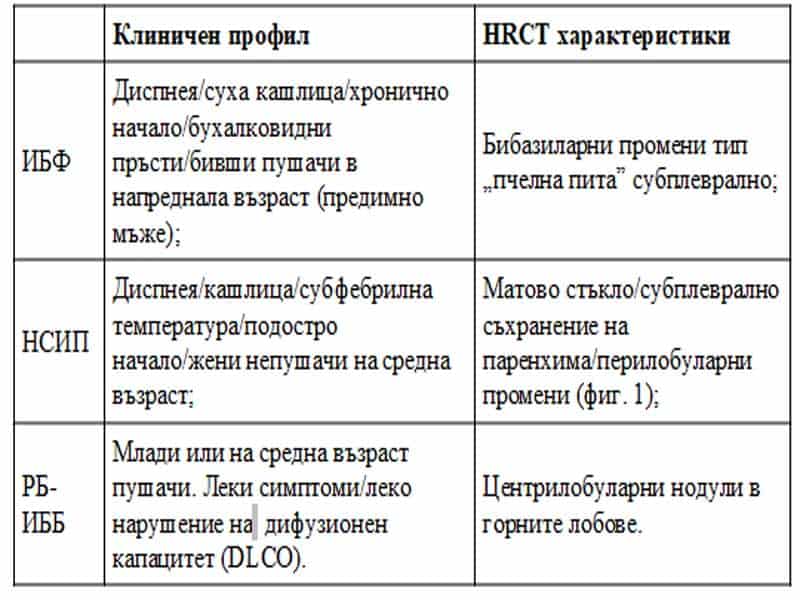
Съществуват множество заболявания, които протичат със симптоми и клинични прояви, наподобяващи ИБФ – повече от 200 белодробни болести и синдроми, което значително затруднява извеждането на прецизна диагноза. Повечето са с хроничен ход и пациентите се представят с дискретни или нетипични симптоми, ограничени предимно в белите дробове. При по-малка част от случаите симптомите бързо прогресират и водят до дихателна недостатъчност. Понастоящем се счита, че комбинацията от клинични признаци, лабораторни тестове и характерен образ на HRCT има диагностична стойност при > 50% от пациентите в групата идиопатични болести (табл. 5)25. За постигане на този успех се налага стриктно спазване на основните диагностични стъпки за изработване на правилен диференциално-диагностичен план.
Табл. 5 Идиопатични интерстициални пневмонии: типични клинико-радиологични аспекти
Мултидисциплинарен подход
За поставяне на точна диагноза е необходимо да се прилага мултидисциплинарният подход. Необходима е тясна комуникация между клиницист, рентгенолог и патолог, с обсъждане между тях на всички клинични данни (професионални вредности, тютюнопушене, придружаващи заболявания, функционални показатели на дишането, лабораторни изследвания), рентгенологични промени и морфологични характеристики.
Диагностичен алгоритъм при ИБФ
ИБФ се среща най-често при възрастни пациенти над 60 години, обикновено мъже, пушачи, при които са изключени всички други възможни етиологични фактори, водещи до развитие на прогресивна интерстициална белодробна фиброза – експозиция на вредни пулмотропни вещества от околната среда и в бита; медикаменти с профибротичен ефект, системни съединителнотъканни заболявания и др.
Подробното снемане на анамнезата и обстойния клиничен преглед са първата стъпка в диагностичния процес. Пациентите трябва да бъдат разпитвани подробно за основните рискови фактори при ИБФ, а именно: тютюнопушене, контакт със замърсители на околната среда – метални прахове (азбест, силикоза и др.), инфекциозни причинители (EBV, HCV, CMV, HSV и др.), генетична предиспозиция, ГЕРБ и др. При наличие на специфичните за ИБФ симптоми и клинични находки диагнозата се верифицира след целенасочено провеждане на следните основни диагностични процедури: конвенционална рентгенография на бял дроб, HRCT, функционално изследване на бял дроб (спирометрия, дифузионен капацитет), изследване на артериални кръвни газове или пулсоксиметрия, отворена белодробна биопсия с хистологична верификация, серологични диагностични тестове (при съмнение за системни съединителнотъканни болести), шестминутен тест при ходене(6MWT)26-30
Прогноза и причини за смърт при пациенти с идиопатична белодробна фиброза
ИБФ е прогресиращо, необратимо и смъртоносно заболяване, като продължителността на живота от времето на поставяне на диагноза е средно между 2-5 години 10.
Анализът на причините за смърт при болни с ИБФ показва, че най-честата причина е острата екзацербация на ИБФ (40%), хронична респираторна недостатъчност (24%), пневмонии (7%), кардиоваскуларни болести (7%), други (10%) и неизвестни (5%)31. Необходимо е да се отчита и рискът от развитие на белодробна тромбемболия (БТЕ) при ИБФ, който е с 34% по-висок, отколкото при здравата популация. Болните с ИБФ и БТЕ умират в по-млада възраст спрямо тези само с ИБФ. Освен това рискът за развитие на белодробен рак при ИБФ е 7.31 пъти по-висок в сравнение с общата популация, което най-вероятно е свързано с високата фибротична активност32.
Лечение
Обобщавайки терапевтичното поведение при ИБФ може да се каже, че засега няма установено оптимално, радикално куративно медикаментозно лечение, а то е по-скоро палиативно. Значими са промените във фармакологичното лечение на ИБФ за периода 2000 – 2015 година.
На базата на приключили мултицентрови проучвания през 2015 г. се стигна до нови ръководни препоръки, основната която е силно–не относно използването на комбинацията преднизон, азатиоприн и N-ацетилцистеин и слабо-да за използване на pirfenidone и nintedanib като специфична ИБФ-терапия, с която се постига редуциране на болестната прогресия, намалява степента на влошаване на FVC и смъртността вследствие намаляване епизодите на остра екзацербация33-40..
Pirfenidone
Механизмът на действие на pirfenidone върху фиброобразуването не е напълно изяснен, но се счита, че ефектите му са свързани с TGF-β (transforming growth factor) (протеин, свързан с растежа и фиброобразуването) и TNF-α (проинфламаторен протеин).
Три големи рандомизирани проучвания (CAPACITY 004 и 006 и ASCEND) изследват ефективността на pirfenidone с първична крайна цел – темпа на снижение ФВК 41,42.
Обобщени данни от 3-те проучвания при общо 1247 пациенти докладват, че за 1 година pirfenidone снижава с 43,8% дела на пациентите с ускорена загуба на ФВК и увеличава с 59,3% дела на тези, при които няма ускорена загуба на ФВК43.
Най- често наблюдаваните странични явления са гастроентерологични: гадене (37,6%), диария (28,1%), диспепсия (18,4%), повръщане (15,9%) и кожни: обриви (25,0%) и фоточувствителност. Наблюдавани са още повишение на чернодробните ензими. Увеличение на АЛАТ и/или АСАТ над 3 пъти над нормата е наблюдавано при 3,1% от пациентите, основно в първите 6 месеца от лечението44.
Медикаментът е разрешен за употреба в България и към настоящия момент кандидатства за реимбурсация от НЗОК.
Nintedanib
Nintedanib е троен инхибитор на тирозинкиназни рецептори (тромбоцитен, васкуларен и фибробластен) и на растежни фактори, които участват във фиброгенезата. Приключилото BIBF 1120 проучване даде основание на Европейската комисия да даде разрешение (2015 г.) за използването на медикамента като първа линия на поведение при лечението на ИБФ наред с пирфенидона, като отчита, че той също редуцира болестната прогресия и проявите на остра екзацербация. В две големи рандомизирани, 52-седмични, клинични проучвания (INPULSIS-1 и 2), включващи общо 1066 пациенти, приемащи 150 мг nintedanib дневно или плацебо, се доказва, че nintedanib значително понижава годишното влошаване на ФВК средно с 50% в сравнение с пациентите, които са приемали плацебо. И при nintedanib основните странични явления са гастроентерологичните (диария, гадене, повръщане, намален апетит), назофарингит, диспнея, инфекция на горни дихателни пътища), като някои от тези прояви могат да бъдат тежки 45, 46.
От нефармакологичното лечение белодробната трансплантация е с препоръка силно „ДА“ и дискусията за нея трябва да започне 3–6 месеца след диагностицирането. Ако болният е клинично индициран и няма противопоказания, трябва да се обърне към трансплантационен център и информиране относно риска и ползата от белодробната трансплантация.
Заключение
Въпреки че диагностицирането и лечението на ИБФ остава постоянно предизвикателство дори за най-опитните клиницисти, последните постижения в диагностиката и лечението при пациенти с ИБФ предлагат значителен оптимизъм, че имаме напредък срещу тази болест.
Референции:
1.Иванов, С. Дифузни белодробни фибрози. Монография, София, 2002, 1-261
2.Иванов, С.Идиопатична белодробна фиброза.Белодробни болести. Учебник под редакцията на проф.К.Костов, София, 2016.
3.American Thoracic Society, European Respiratory Society. American Thoracic Society/ European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165:277-304
4. W.D.Travis, U.Costabel, D.M.Hansell et al, An Official American Thoracic Society/European Respiratoty Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias; Am J Respir Crit Care Med 2013; Vol188(6);733-748
5. Raghu G, Rochwerg B, Zhang Y, et al. An Official ATC/ERS/JRS/ALAT Clinical PracticeGuideline: Treatment of idiopathic Pulmonary Fibrosis: Executiv Summary An Update of the 2011 Clinical Practice Guideline AM J Respir Crit Care Med 2015; 192(2):238-248
6. Gribbin J, Hubbard RB, Le Jeune I, et al. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK.Thorax 2006; 61:980-85
7.Raghu G,Weycker D, Edelsberg J, et al.Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006; 174:810-16
8.Raghu G, Collard HR, Egan JJ, et al. An official ATC/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management.Am J Respir Crit Care Med 2011;183:788-824
9.Luppi F, Spagnolo P, Cerri S, Richeldi L. The big clinical trials in idiopathic pulmonary fibrosis. Curr Opin Pulm Med 2012; 18: 428–432
10. J.A. Bjoraker, J.H. Ryu, M.K. Edwin, et al: Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998, 157(1):199-203
11. Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/ fibroblastic cross-talk disorder. Respir Res 2002; 3: 3
12.Raghu G., Collard H.R., Egan J.J., et al. On behalf of the ATS/ERS/JRS/ALAT Committee An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management Am J Respir Crit Care Med 2011 Vol 183. pp 788–824.
13.Wells AU. The revised ATS/ERS/JRS/ALAT diagnostic criteria for idiopathic pulmonary fibrosis (IPF) – practical implications. Respir Res. 2013;14 Suppl 1:S2.
14. Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015;192(2):e3-e19.
15. Sundaram, B.H.Gross, F. Martinez, et al: Accuracy of high-resolution CT in the diagnosis of diffuse lung disease: effect of predominance and distribution of findings. AJR Am J Roentgenol 2008, 191(4):1032-9
16. M.D. Martin, J.H. Chung, J.P. Kanne; Idiopathic Pulmonary Fibrosis; J Thorac Imaging 2016;00:000–000
17. A.U. Wells;The revised ATS/ERS/JRS/ALAT diagnostic criteria for idiopathic pulmonary fibrosis (IPF) – practical implications; Wells Respiratory Research 2013, 14; Suppl1:S2
18. Raghu G, King TE Jr, Bradford WZ, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2011;183:788–824.
19 .Poletti V, Ravaglia C, Gurioli C, et al. Invasive diagnostic techniques in idiopathic interstitial pneumonias. Respirology 2016; 21: 44–50.
20. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society Statement: update of the International Multidisciplinary Classification of Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013; 188: 733–48
21. Poletti V, Ravaglia C, Gurioli C, et al. Invasive diagnostic techniques in idiopathic interstitial pneumonias. Respirology 2016; 21: 44–50.
22. Poletti V, Casoni GL, Gurioli G, et al. Lung cryobiopsies: a paradigm shift in diagnostic bronchoscopy.
Respirology 2014; 19: 645–54
23. Casoni GL, Tomassetti S, Cavazza A, et al. Transbronchial lung cryobiopsy in the diagnosis of fibrotic interstitial lung diseases. PLoS ONE 2014; 9: e86716.
24. Annema JT, Rabe KF. State of the art lectures: EUS and EBUS in pulmonary medicine. Endoscopy 2006; 38: S118–22.
25. Jacob J, Hansell DM. HRCT of fibrosing lung disease. Respirology 2015; 20: 859–72.
26. Wim A. Wuyts, Cavazza А., Rossi G., et al. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev 2014; 23: 308–319. – December 01, 2014
27. Idiopathic pulmonary fifibrosis overview NICE 2016 – National Institute for Health and Care Exellence.
28. Kawano-Dourado L, Kairalla RA. Usual interstitial pneumonia: a pattern or a disease? A reflection upon the topic. J Bras Pneumol. 2013;39(1):111-2.
29. Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: What is the effect of a multidisciplinary approach to diagnosis? Am J Respir Criti Care Med 2004;170:904–10.
30. Wilson K, Raghu G. The 2015 guidelines for idiopathic pulmonary fibrosis: an important chapter in the evolution of the management of patients with IPF Eur Resp J 2015;44; 4: 883 – 886.
31. Günther A, Korfel M, Mahavadi P, et al. Untravelling the progressive pathophysiology of idiopatic pulmonary fibrosis. Eur. Respir. Rev. 2012; 21, 124, 152-160.
32. Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med. 2000; 161(1):5-8.
33.Noble PW, Albera C., Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet 2011; 377: 1760-1769.
34.Hilberg O, Simonsen Ulf, du Bois R, et al. Pirfenidone: significant treatment effects in idiopathic pulmonary fibrosis. Clin, Respir. J – 2012; 131-142.
35. Raghu G, Anstrom KJ, King TE Jr, et al. Prednisone, azathioprine and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366; 1968-1977.
36. Noble P, Albera C, Bradford W, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016, 47, 243-253.
37. Currow, D. The inpulsis trial of idiopathic pulmonary fibrosis treatment: explaining further discrepancies on exacerbation. Eur Respir J. 2016, 47, 342-343.
38. Milder, K. Swizching to Nintedanib after discontinuation of Pirfenidone due to adverse events in IPF. Eur Respir J. 2015; 46: 1217-1221.
39. Cottin V, W. Wuyts. Insights into idiopathic pulmonary fibrosis in the real world. Eur Respir J 2015; 46: 16-18.
40. Collard H., W. Bradford, V. cottin, et al. A new era in idiopathic pulmonary fibrosis: considerations for future clinical trials. Eur Respir J 2015; 46: 186-196.
41. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. The Lancet. 2011;377(9779):1760-1769
42. King TE, Bradford WZ, Castro-Bernardini S, et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. New England Journal of Medicine. 2014;370(22):2083-2092.
43. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47(1):243-253.
44. Lancaster L, Albera C, Bradford WZ, et al. Safety of pirfenidone in patients with idiopathic pulmonary fibrosis: integrated analysis of cumulative data from 5 clinical trials. BMJ Open Respiratory Research. 2016;3(1):e000105.
45. Neil, A. C. New Therapeutic Targets in Idiopathic Pulmunary Fibrosis. Am J. Respir Crit Care Med 190, 2014, 867-878.
46. Raghu, G. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary fibrosis: Evidence – based Guidelines for Diagnosis and Management. Am J Respir Crit Care Med 2011; 183, 788-834.
Авторски колектив: Д-р Дарина Стоянова, Доц. Наталия Стоева
Отделение по пневмология
МБАЛ „Токуда Болница София“, гр.София ул.Н.Вапцаров номер 51Б
Тел.: 0886249064 – Д-р Д.Стоянова



